Jun 24, 2025
Articles
What is CAPA and Why It's Important? [Comprehensive Guide]

Martin Ramirez

Regulatory bodies, such as the FDA, expect manufacturers to have effective CAPA systems that not only correct issues but also prevent them from recurring, while maintaining detailed documentation and accountability.
With increasing scrutiny and compliance requirements, having a detailed, risk-based CAPA process is more important than ever.
In this guide, we’ll cover what is CAPA, explore common pitfalls, and share best practices for building a system that’s audit-ready and aligned with today’s regulatory expectations.
Let’s get started.
What is CAPA?
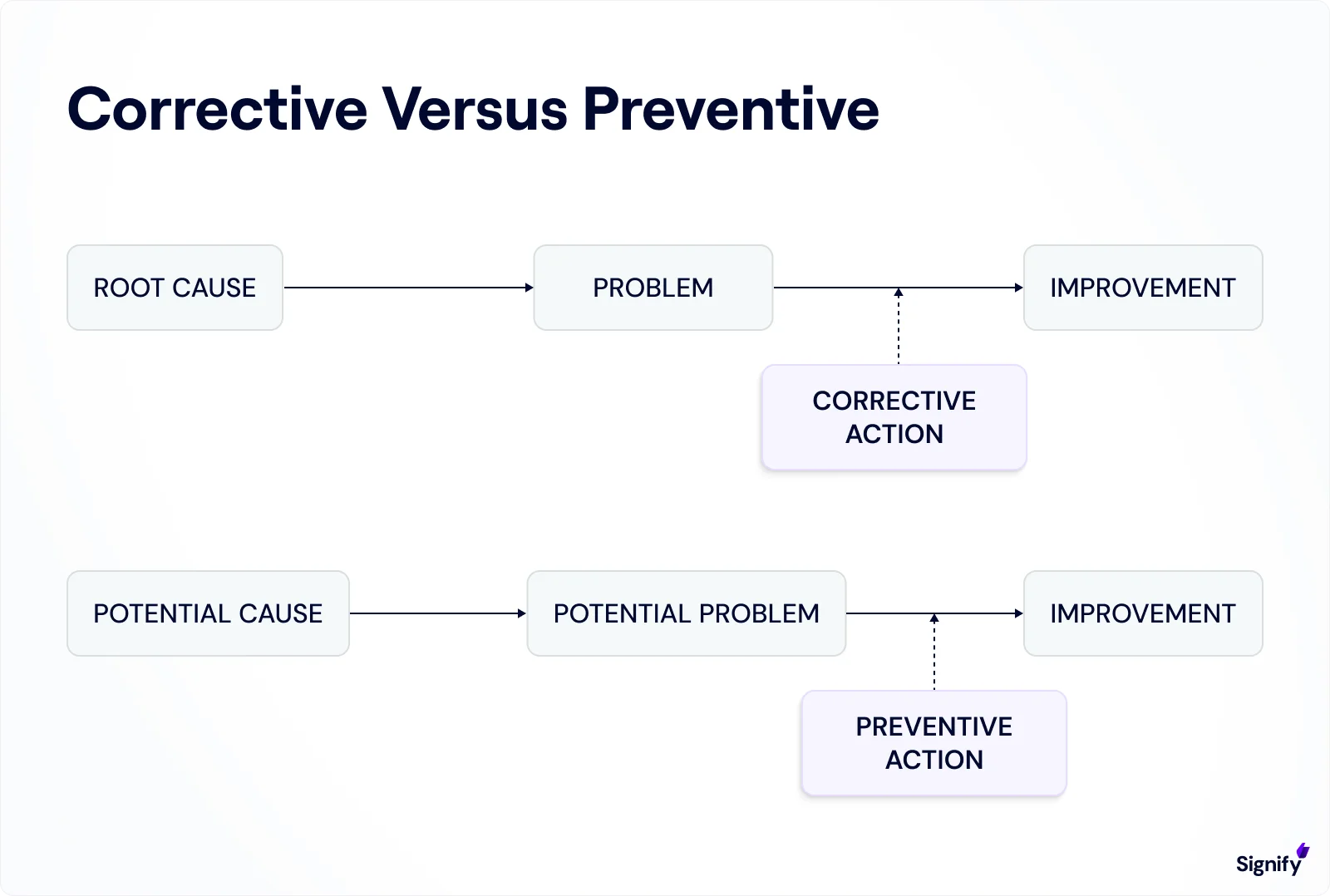
CAPA stands for Corrective and Preventive Action.
It became a regulatory requirement through systems like the FDA’s Quality System Regulation (21 CFR Part 820) and ISO 13485.
Over the years, it evolved into a key process in ensuring product quality, patient safety, and regulatory compliance.
It’s a fundamental process used in regulated industries to identify, investigate, and fix problems in a way that prevents them from recurring or happening in the first place.
➤ The Two Components of CAPA:
Corrective Action addresses existing issues. For example, fixing a non-conforming batch of products.
Preventive Action is proactive. It identifies potential issues before they occur and takes steps to prevent them, such as improving training to avoid future deviations.
How CAPA Supports Compliance in 2025
Regulators today expect a transparent, traceable record of how issues are handled.
CAPA provides that structure, especially in digital systems where audit trails are mandatory.
➤ The Role of CAPA in FDA, ISO 13485, and EU GMP Compliance
CAPA is referenced in several major regulatory frameworks:
FDA: 21 CFR Part 820.100 requires establishing and maintaining Corrective and Preventive Action (CAPA) procedures.
ISO 13485: Clauses 8.5.2 and 8.5.3.
EU GMP: Chapter 1.4 emphasizes the identification of root causes and the taking of preventive actions.
When Should CAPA Be Initiated?
A well-timed CAPA can prevent future failures, protect users, and satisfy regulatory expectations, but using it indiscriminately can backfire.
Key Events That Trigger CAPA Procedures
In practice, CAPAs should be initiated when clear quality risks emerge.
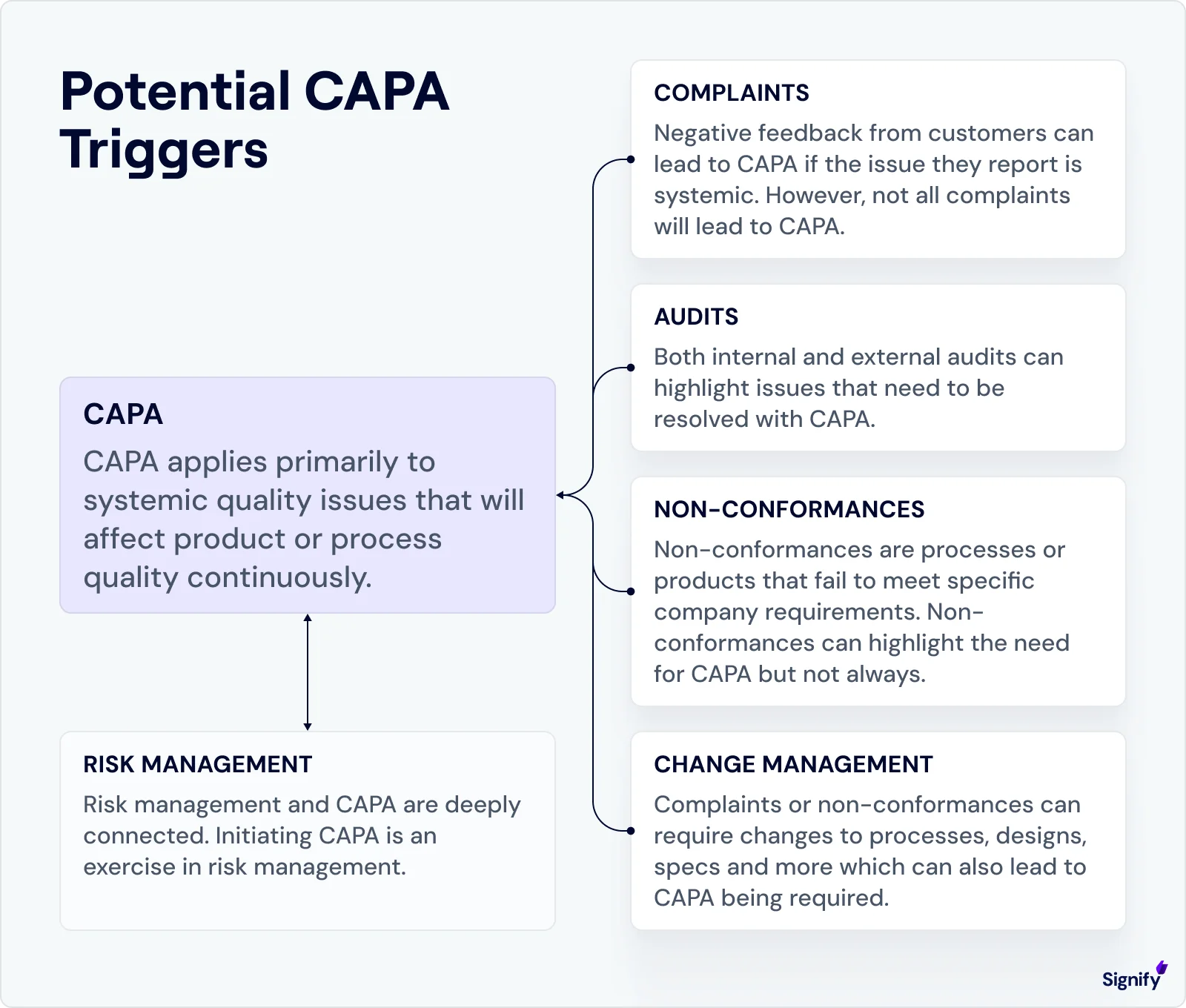
➤ Common scenarios include:
Nonconforming product reports reveal recurring issues. If several units are found to be out of specification, and the cause can be traced back to a systemic issue, such as a miscalibrated machine, a CAPA should be opened to investigate the root cause and prevent recurrence.
Multiple complaints about the same defect or a serious adverse event. A cluster of customer complaints pointing to the same flaw, or even a single critical event (like an injury or serious malfunction), can justify immediate CAPA action due to the potential risk involved.
Audit findings point to systemic breakdowns. Whether during internal audits, supplier evaluations, or regulatory inspections, repeated deficiencies are often observed. For example, inconsistent record-keeping may signal the need for corrective and preventive measures.
Statistical trends exceed control thresholds. If your production, quality control, or complaint metrics show an upward trend or cross internal warning limits, a preventive CAPA can help address the issue before it escalates.
Management review flags recurring risks. Senior leaders may notice broader trends during review meetings, such as rising scrap rates or an increasing backlog of unresolved CAPAs, prompting investigation and formal action.
The Risk of Overusing or Underusing CAPA
Finding the right balance is essential.
➤ Many companies fall into one of two traps:
Underuse: Failing to open a CAPA when one is warranted can lead to repeat incidents and regulatory risk. Issues go unaddressed, the root cause remains, and future failures become more likely. Regulatory bodies view this as a serious oversight, particularly when safety or compliance is at stake.
Overuse: On the other end of the spectrum, being “CAPA-happy” by launching a CAPA for every minor deviation or complaint drains resources. It creates administrative overhead, clogs workflows, and causes key investigations to stall. When everything becomes a CAPA, nothing gets the attention it deserves.
To avoid both extremes, always ask:
➤ Is this a systemic or high-risk issue?
If yes, open a CAPA. If not, consider resolving the issue through existing quality processes, such as deviation handling, change control, or retraining.
Using a Risk-Based Approach to Determine CAPA Need
Every CAPA decision should begin with risk assessment.
Focus your efforts on problems that are likely to recur or carry significant impact.
A simple way to evaluate the need for CAPA is to ask:
Did the incident result in, or could it reasonably result in, serious harm, injury, or product failure?
Is this issue part of a pattern? Recurrence often indicates deeper systemic failure.
Does your risk matrix rank this issue as high severity or high probability?
Even a one-time event may merit a CAPA if the stakes are high enough (e.g., patient safety, regulatory exposure, or major operational disruption).
The goal is to use CAPA strategically, not reactively, directing your investigation and improvement efforts where they will have the greatest impact.
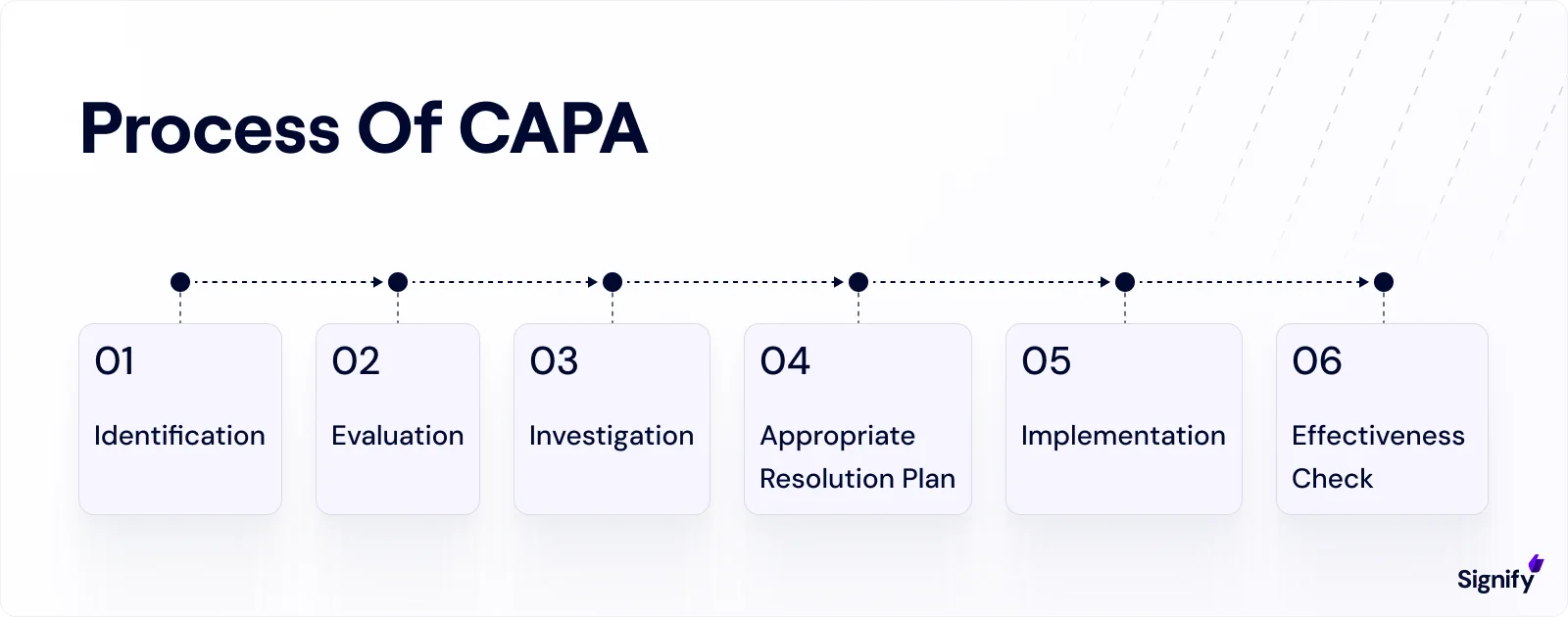
Core Components of an Effective CAPA System
A strong CAPA system follows a clear, structured process from problem identification to verification of effectiveness.
Each step must be handled carefully, with consistent documentation and input from multiple functional areas.

1. Problem Identification and Documentation
Every CAPA starts with a precise and well-documented problem statement.
➤ This means capturing:
What happened,
When and where it occurred,
Who was involved,
The defect or issue observed,
Any supporting data or test results.
For example, if a machine starts producing defective parts, your report should include defect rates, machine logs, shift records, and any immediate containment steps taken.
Poor documentation leads to confusion and weak investigations, while a clear paper trail ensures consistency and readiness for audits later.
2. Conducting a Risk Assessment
Once the issue is documented, evaluate the risk to determine the best course of action.
➤ Use tools such as:
A basic risk matrix
More advanced tools
Score the problem based on severity, frequency, and detectability. If the risk is low, a quick fix may be a suitable solution.
If it’s medium to high, a full CAPA should be initiated.
Risk assessment also helps justify resource allocation and timelines within your CAPA plan.
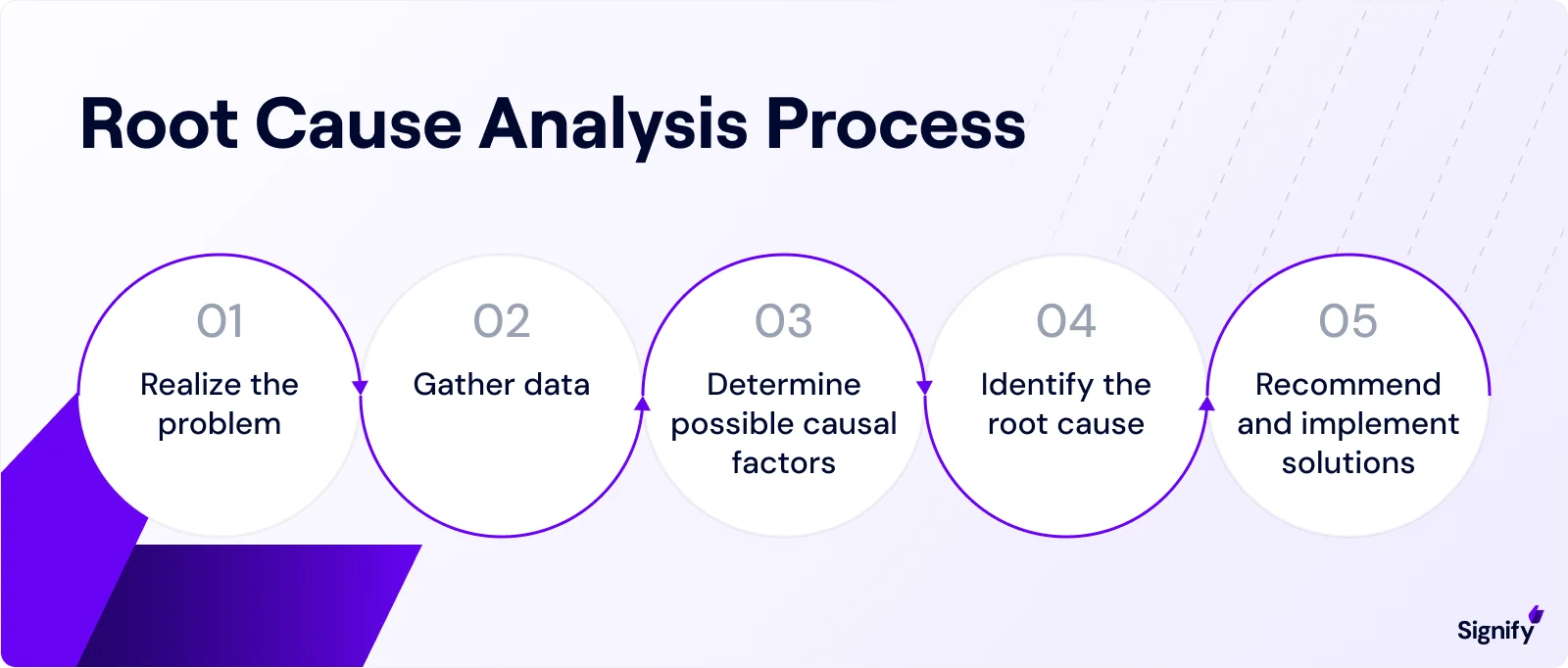
3. Root Cause Analysis Tools and Techniques
Identifying the real root cause is essential.
➤ Use appropriate RCA tools depending on the issue’s complexity:
5 Whys: Simple, fast, best for straightforward problems.
Fishbone (Ishikawa) Diagrams: A visual tool to group causes under categories like People, Equipment, or Methods.
Pareto Charts: Highlight the top contributors to recurring issues.
FMEA: Systematically assesses failure modes, effects, and prioritization via RPN.
Fault Tree Analysis: Logical mapping of failure events from the top down.
Also, always involve a cross-functional team, like quality, engineering, manufacturing, and others, so the analysis isn’t siloed or superficial.
Avoid attributing errors to “human error” unless supported by deeper systemic findings (e.g., training gaps or unclear Standard Operating Procedures (SOPs).
4. Creating and Executing a Corrective and Preventive Action Plan
With the root cause identified, outline clear and actionable steps to resolve the issue and prevent recurrence.
➤ A solid action plan includes:
Defined tasks,
Assigned owners,
Due dates,
Linked root causes.
Typical CAPA actions may include retraining staff, revising standard operating procedures (SOPs), implementing additional inspections, changing suppliers, modifying equipment, or redesigning processes to address the issue.
Each action must address the actual cause, not just the symptom, and ensure that detailed records are kept of what was done, when, and by whom.
5. Verifying and Validating CAPA Effectiveness
Once actions are implemented, you must confirm they worked.
➤ This involves:
Reviewing relevant metrics or KPIs.
Retesting affected products or processes.
Auditing updated procedures or systems.
Gathering evidence of training effectiveness.
CAPA is not complete until effectiveness is proven and documented.
If the problem returns or only partially resolves, the CAPA should be revised or reopened.
Regulators, including FDA inspectors and ISO auditors, will verify this step during audits.
6. Documentation and Audit Readiness
Thorough documentation is crucial for ensuring compliance and maintaining traceability.
➤ You should maintain records of:
The initial problem report,
Risk assessments,
Investigation findings,
RCA diagrams,
Action plans and completion logs,
Effectiveness checks,
Final closure approvals.
Good documentation ensures audit readiness and creates organizational learning.
Regulators expect a thorough history of how the issue was handled, and without it, your CAPA may fail, even if the technical issue has been resolved.
Best Practices for CAPA Implementation
CAPA plays a central role in identifying, resolving, and preventing quality issues.
Today, effective systems rely on digital tools, structured planning, and continuous feedback to ensure lasting improvements.
Use Digital Tools
Modern CAPA systems are built on AI quality management platforms that automate workflows, enforce consistency, and maintain secure records.
➤ These systems:
Provide structured workflows and mandatory fields to ensure completeness.
Generate real-time dashboards and alerts for pending actions.
Create comprehensive audit trails with timestamps and electronic signatures.
Link CAPA to related processes, such as FMEA, change control, complaints, and audits.

Support collaboration across departments.
Most of the tools also integrate RCA methods, analytics, and trend detection to support smarter decision-making.
Set Realistic Timelines and Assign Clear Responsibilities
Poorly managed CAPAs often fail due to vague ownership or unachievable deadlines.
➤ Avoid this by:
Assigning a clear owner to each CAPA task (e.g., root cause analysis, corrective action, effectiveness check).
Using structured role frameworks (such as RACI) for accountability.
Setting realistic timelines based on task complexity.
Reviewing progress regularly in team meetings or management reviews.
Leveraging digital tools to flag overdue actions and keep plans moving.
Align CAPA With Continuous Improvement (CI) Goals
CAPA shouldn’t operate in a vacuum, as it aligns with the broader cycle of continuous improvement and should contribute to long-term quality objectives.
➤ For example:
Treat each CAPA as part of the PDCA (Plan-Do-Check-Act) cycle.
Use recurring issues to trigger Lean or Six Sigma improvement initiatives.
Tie CAPA outcomes to strategic KPIs (e.g., reduced defect rates, improved training outcomes).
Review completed CAPAs for insights and trends during management reviews to inform future actions and decisions.
Signify’s AI Compliance Agent: Smarter, Faster CAPA Management
Signify is an AI compliance agent that helps manufacturers detect, investigate, and resolve quality issues with speed and precision.
Its AI compliance agents automate workflows and ensure every step meets regulatory standards, all within a secure, traceable system.
Here is how we assist:
Workflow Automation and Speed: Signify’s AI agents automate compliance workflows, reducing manual effort and accelerating approvals. CAPA investigations, documentation, and follow-up actions are now completed within hours, minimizing delays and enabling faster responses to quality issues.
AI-Driven Risk Identification: Signify’s AI automatically detects issues and evaluates the risk level of designs, documents, and records.
Smart File Management and Traceability: All files and records are cross-referenced and traceable. The AI builds a dynamic traceability matrix, making it easier to demonstrate effectiveness and compliance during audits.
Audit Trails and Documentation: Every action and change is time-stamped and locked in a secure audit trail.
Automated Compliance Review: AI agents continuously validate that activities align with internal procedures and global regulations. This reduces the risk of missing documentation steps and supports ongoing compliance.
Extending Compliance Across Industries:
Furniture
Tools & DIY Equipment
Try Signify for free today and see how it can automate your process, reduce quality risks, and keep your organization audit-ready and compliant with global regulations.
Jun 24, 2025