Jun 17, 2025
Articles
Understanding the Regulatory Framework for OTC Drugs

Martin Ramirez

Did you know that the OTC Pharmaceuticals market worldwide is projected to generate a revenue of US$211.81 billion in 2025, with an anticipated annual growth rate of 4.84% (CAGR) from 2025 to 2029?
As someone navigating the OTC drugs industry, you’ve likely encountered the complexities of regulatory compliance but may still be unsure how it impacts your business.
In this article, we’ll break down the essential steps to help you understand and navigate the regulatory framework for OTC drugs.
Let’s dive in!
What Are Over-the-Counter (OTC) Drugs?
Over-the-counter (OTC) drugs play a vital role in the U.S. healthcare system, enabling consumers to treat common ailments without a prescription.
These drugs are available in stores and online and can be purchased directly by consumers.
The FDA defines OTC drugs as those that "do NOT require a doctor’s prescription" and are typically used to treat common conditions in an otherwise healthy population.
Here’s a breakdown of the essential aspects of OTC drugs:
Self-Selection and Use: OTC drugs allow individuals to self-select and treat symptoms like pain, cough, allergy, or heartburn, as long as they follow the label directions.
FDA Evaluation: The FDA evaluates these drugs to ensure they are safe for over-the-counter use and effective in treating the conditions for which they are intended.
➤ Common OTC Drug Examples:
Pain relievers (like acetaminophen or ibuprofen)
Cold and allergy remedies
Antacids for heartburn
Topical creams for skin conditions
Understanding this framework helps consumers, retailers, and manufacturers know how OTC medicines are regulated and how they stay on store shelves.
➤ Important to Know:
The FDA oversees OTC drugs through specific pathways and standards to ensure these widely available treatments are used safely without the need for direct supervision by a healthcare provider.
Key Differences Between OTC and Prescription Medications
Although OTC and prescription drugs both treat health conditions, there are important differences in how they are regulated, used, and distributed.
The table below compares the two types side-by-side:
Feature | OTC Drugs | Prescription Drugs |
Requirements | Can be purchased without a doctor’s order. | Require a valid prescription from a licensed healthcare provider. |
Availability | Sold directly “over the counter” in pharmacies, grocery stores, and online. | Available only at pharmacies; often located behind a counter or locked; must be dispensed by a pharmacist. |
Intended Use | Intended for self-treatment of minor or common conditions (e.g., headache, cold). | Treat more serious or complex conditions; use under physician supervision. |
FDA Regulation | Regulated under OTC drug monographs or an NDA; labeling must follow the standard “Drug Facts” format. | Regulated through the New Drug Application (NDA) process; labels may have prescriber instructions. |
The FDA highlights the key differences between prescription and over-the-counter (OTC) drugs, noting that prescription drugs require a doctor’s prescription and undergo the New Drug Application (NDA) approval process.
In contrast, OTC drugs do not require a prescription and generally follow the OTC monograph system, unless special approval is needed.
Regulatory Framework for OTC Drugs: FDA Oversight
The FDA oversees all drug products, including OTC drugs, under the Federal Food, Drug, and Cosmetic Act (FD&C Act).
The FDA’s Center for Drug Evaluation and Research (CDER) ensures OTC drugs are safe and effective, handling pre-market approval and post-market surveillance.
The Role of the FDA in Regulating OTC Drugs
The FDA’s role is to establish safety and efficacy standards for OTC medications, evaluate products against these standards, and enforce compliance.
This includes:
Establishing Standards (Monographs): The FDA defines active ingredients, doses, formulations, and labeling for OTC drugs in monographs, which serve as a "recipe book" for approved products.
Reviewing Non-Monograph Products (NDAs): The FDA reviews NDAs or ANDAs for new OTC drugs with unapproved ingredients or uses to ensure safe, effective self-use.
Setting Labeling Requirements: The FDA mandates a standardized “Drug Facts” label format for all over-the-counter (OTC) drugs, ensuring that key information is easily accessible and understandable.
Monitoring Manufacturing (cGMP): The FDA requires OTC drug manufacturers to follow Current Good Manufacturing Practices (cGMP), as outlined in 21 CFR Parts 210 and 211, ensuring that products are consistently of high quality, similar to those of prescription drugs.
Post-Market Surveillance: The FDA monitors safety through adverse event reporting, inspections, and consumer complaints, taking action when issues such as side effects or contamination arise.
Under the FDA framework, both OTC and prescription drugs must meet the same basic safety standards, though their review processes differ.
Also, the FDA requires that both OTC monograph and NDA products meet safety and effectiveness standards, be manufactured under Current Good Manufacturing Practice (cGMP) conditions, and be properly labeled.
Two Main Approval Pathways for OTC Drugs
There are two primary pathways for an OTC drug to be legally marketed in the United States:
➤ OTC Monograph Process:
Many common over-the-counter (OTC) ingredients are approved under FDA regulations known as "OTC drug monographs" (21 CFR Part 330).
Products containing monograph-approved ingredients, doses, formulations, and labeling automatically qualify as Generally Recognized as Safe (GRAS) and can be marketed without additional FDA pre-approval.
➤ New Drug Application (NDA) or Abbreviated NDA:
If an OTC drug doesn’t fit within an existing monograph (e.g., new ingredient, dose, or indication), it must go through the NDA process, including submitting clinical and safety data.
Once approved, the drug can be marketed only with the specific formulation and labeling approved by the FDA. This includes both new OTC drugs and Rx-to-OTC switches.
➨ To summarize, the FDA offers two routes for OTC drugs:
Products that meet monograph standards can be marketed without further clearance.
Products that do not fit an existing monograph must undergo New Drug Application (NDA) review, ensuring safety, efficacy, and proper consumer information.
Pro Tip
Signify’s Regulatory Monitoring keeps you informed of changes to relevant laws (FDA, USDA, ISO, and over 1000 frameworks) and standards in real time.
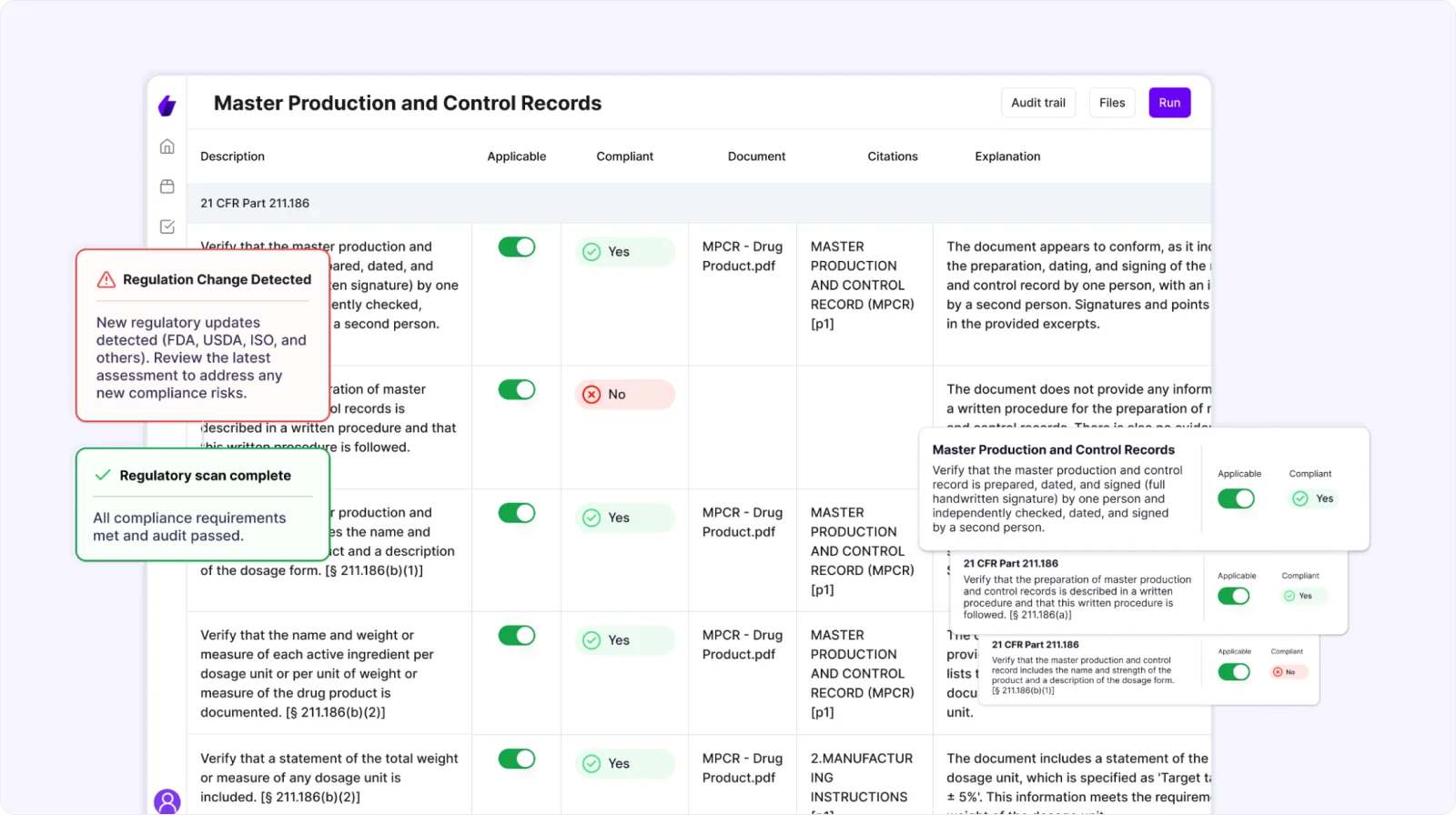
Make it a habit to review these alerts weekly so you can proactively adapt your compliance documentation and avoid costly disruptions or non-compliance risks.
FDA OTC Drug Labeling Requirements
All nonprescription drug products sold in the U.S. must carry a standardized Drug Facts label, as mandated by the FDA (21 CFR 201.66).
This label ensures that consumers can easily read and compare over-the-counter (OTC) medicines.
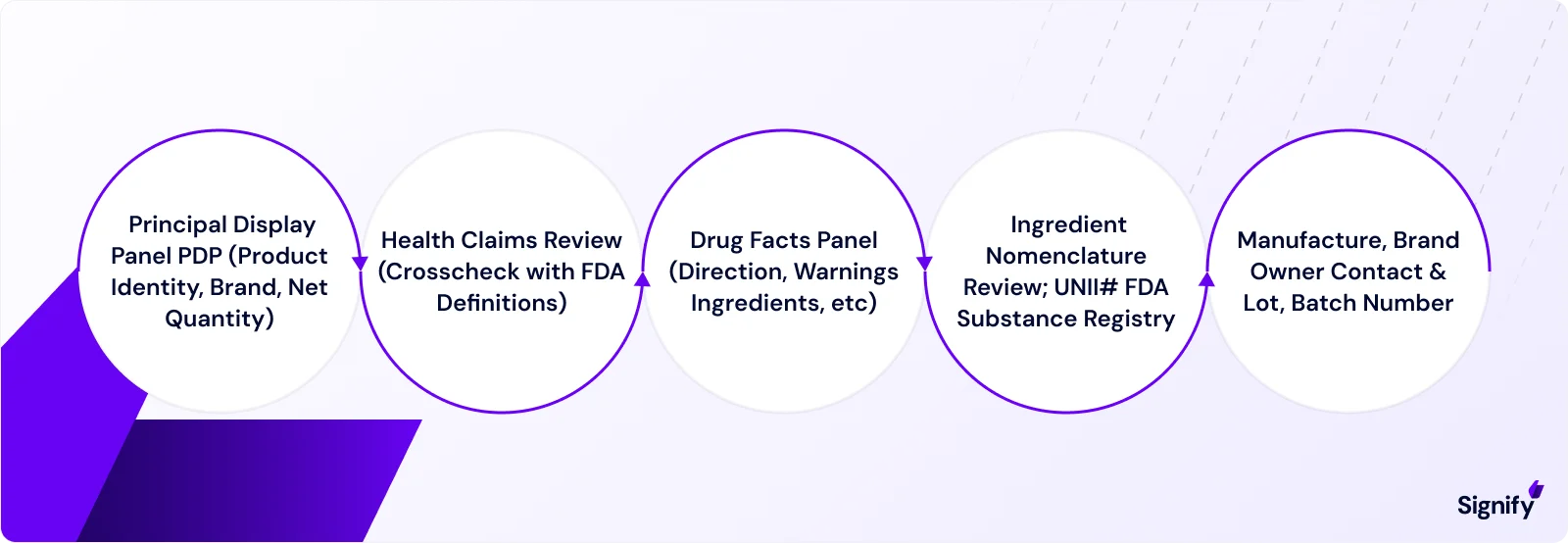
Key aspects of the label include:
➤ Title and Headings:
The label must begin with "Drug Facts" in bold and italic, followed by section headings in bold and subheadings in bold (e.g., “Active Ingredient,” “Purpose”).
All text must be legible, with a minimum font size of 6-point for body text and 8-point for headings.
➤ Active Ingredients:
The active ingredients must be listed by their established name and the amount per dosage unit (e.g., "Acetaminophen 325 mg"). Amounts are specified according to the dosage form (e.g., tablet, teaspoonful).
The active ingredient list must be left-justified and aligned with the "Purpose" section.
➤ Purpose:
This section describes the drug’s general pharmacologic action or category (e.g., "Pain reliever/fever reducer"). If a monograph exists, its term must be used.
For multiple active ingredients with the same purpose, a single description may suffice.
➤ Uses:
Lists the specific symptoms or conditions the product is meant to treat or prevent (e.g., "For relief of headache, minor aches, and pains").
Non-drug uses, like cosmetic or dietary claims, are excluded.
➤ Warnings:
This section includes required cautionary statements about when not to use the product, potential interactions, and special warnings (e.g., pregnancy or breastfeeding).
Warnings must be stated exactly as required to avoid misbranding.
➤ Directions (Dosage Instructions):
Provides dosage instructions, including the amount to take, frequency, and any special considerations.
For multiple age groups, a chart format is used.
Single-step instructions (e.g., "Shake well before use") should be listed as bullets.
➤ Inactive Ingredients:
Lists all non-drug components (e.g., binders, fillers, colors) in alphabetical order.
This helps consumers check for known allergens or sensitivities.
➤ Expiration Date:
The label must include an expiration date to ensure the product remains at its full potency and stability.
If no expiration date is provided, the FDA recommends assuming the product expires three years after manufacture.
➤ Lot/Control Number:
It must be included to identify the manufacturing batch. This is crucial for traceability and recalls.
➤ Manufacturer (Name & Address):
The label must display the name and address of the manufacturer, packer, or distributor.
If this is not clearly shown, the product is considered misbranded.
➤ Tamper-Evident Packaging:
Most OTC drugs must be in tamper-evident packaging, with clear labeling stating the feature.
An example could be: “Do not use if printed seal under cap is broken or missing.”
➤ Label Updates for Formulation or Safety Changes:
OTC labels must be kept up-to-date. If a product's formulation or safety information changes, the label must be revised.
For products marketed under a New Drug Application (NDA) or Abbreviated New Drug Application (ANDA), manufacturers must submit supplements for any major changes.
Formatting and Compliance Guidelines
The FDA sets strict guidelines for how the label must be formatted:
➤ Font Size and Readability:
"Drug Facts" must be the largest text on the label, and headings must be at least 8-point type.
The body text should be at least 6-point to ensure readability.
➤ Bullets and Lists:
Warnings and Directions sections must use bullet points for clarity.
For age-based dosing, a table with horizontal lines is required.
➤ Panel Continuation:
If the Drug Facts content spans multiple panels, each continuation panel must be labeled “Drug Facts (continued)” with a hairline border.
Pro Tip
Signify’s Artwork Validation instantly checks product labels and packaging against current regulations, helping you catch errors before they escalate.
Use it early in your product development process to minimize costly redesigns and ensure every label is market-ready from day one.
The OTC Drug Monograph System: A Key Regulatory Pathway
OTC drug manufacturers must adhere to the same Current Good Manufacturing Practice (cGMP) regulations as prescription drug manufacturers, outlined in 21 CFR Parts 210 and 211, to ensure consistent product quality..
cGMP is not just a single practice but a suite of requirements that together ensure drug quality.
Key elements include:
Quality Control Unit: A qualified unit must oversee quality, approve procedures, and have the authority to hold or reject products failing quality tests (21 CFR 211.22).
Raw Material Testing: All raw materials must be tested or certified before use to ensure quality and prevent contamination (21 CFR 211.84).
Process Validation: Critical manufacturing processes must be validated to ensure consistent product quality, with re-validation for process changes.
In-Process Controls: Various checks are performed during production (e.g., weight, temperature) to ensure consistency; corrective actions are taken for deviations.
Laboratory Testing: Finished batches must be tested for safety and potency before release, including microbial testing for non-sterile products.
Packaging and Labeling Controls: Regulations ensure correct labeling, tamper-evident packaging, and expiration dates (21 CFR 211.132 and 211.137).
Record-keeping: All manufacturing steps must be thoroughly documented, creating a comprehensive history of each batch for traceability purposes.
How the ACNU Rule Impacts the OTC Drug Market
The FDA's Additional Conditions for Nonprescription Use (ACNU) rule, finalized in December 2024, establishes a new pathway for certain drugs to become over-the-counter (OTC) with added safeguards, such as digital questionnaires or mobile apps, to ensure safe use without a prescription.

Under the rule, drugs with ACNUs can be marketed OTC if the sponsor meets these conditions and reports any failures.
➤ Implications for Consumers:
ACNU allows easier access to some medications without a doctor’s visit, but may require consumers to complete additional steps, such as filling out a digital questionnaire or using a QR code.
Concerns include accessibility issues for older adults, those without internet access, and others with limited resources.
➤ Implications for Retailers:
Retailers must verify compliance with ACNU conditions, which may involve scanning QR codes or checking printed forms.
ACNU products could open up new markets, but retailers will need to train their staff and update their systems to manage these changes.
FDA Drug Establishment Registration and Listing Requirements
Apart from product approval, the FDA requires drug companies (including OTC manufacturers) to register their manufacturing establishments and list their products.
Establishment Registration
Establishment registration is mandatory for any facility that manufactures, repackages, relabels, or imports drug products in the United States (with some exceptions).
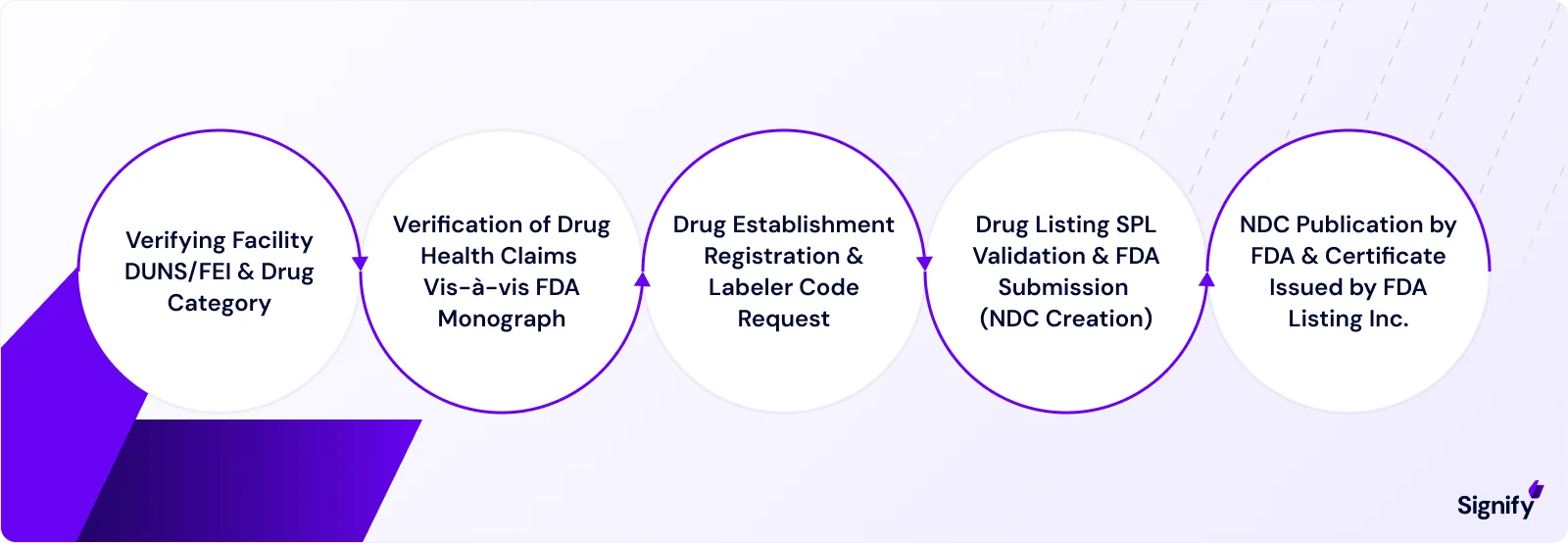
➤ Key requirements include:
Facility Details: Name, address, operations, and contact information.
Foreign Manufacturers: Must designate a U.S. agent.
Timing: Registration must be completed within 5 days of distributing a drug in the U.S.
Annual Renewal: Registration must be renewed annually from October to December.
Product Listing and NDC Assignment
After registering, manufacturers must list their products with the FDA, providing detailed information about each drug.
This process assigns a National Drug Code (NDC) to each product, ensuring accurate tracking and identification of the product.
➤ The NDC consists of three parts:
Labeler Code (assigned to the company)
Product Code (identifies the strength/form)
Package Code (identifies package size/type)

➤ Important aspects include:
Drug Information: Includes proprietary name, dosage, strength, and NDC.
OTC Products: The NDC appears as a barcode on OTC product labels.
Deactivation: If no products are listed under a labeler code for two years, the code is deactivated.
Streamline OTC Drugs Manufacturing Compliance with Signify
Signify is an AI compliance agent designed to streamline pharmaceutical manufacturing operations by ensuring adherence to evolving regulations, minimizing risks, and accelerating product approvals.
With its intelligent platform, Signify continuously monitors global regulatory updates, proactively identifying new requirements and transforming them into actionable, tailored checklists for your products and markets.
➤ Key features include:
Automated Conformity Assessments: Signify conducts real-time assessments to verify that product documentation, labeling, and policies meet all applicable regulations.
Regulatory Risk Tracker: It tracks compliance risks and identifies potential non-conformities before they cause delays or scrutiny.
Smart Compliance Checklists: Custom AI-powered checklists help eliminate confusion and track progress efficiently, ensuring alignment with ever-changing industry standards.
Real-Time Compliance Monitoring: Signify automatically reviews documents to uncover compliance risks, removing the need for manual gap analysis.
Ready to simplify compliance with OTC drug regulations and reduce risks with AI-powered efficiency?
Try Signify today for free and see how easy managing OTC drug compliance can be!
Jun 17, 2025