Dec 10, 2025
Articles
Biosimilar Promotional Labeling Guidance Is Final

Martín Ramírez

Regulatory Standards in Biosimilar Promotional Labeling: What You Need to Know
In a landmark move for the biologics industry, the U.S. Food and Drug Administration (FDA) has finalized its long-awaited guidance on promotional labeling and advertising for prescription biological products. This comprehensive document establishes clear, enforceable boundaries for how manufacturers can market biosimilars, interchangeable biosimilars, and their reference products. Published in the Federal Register on December 10, 2025, the guidance marks the culmination of a regulatory journey that began with draft guidance in 202 and underwent significant revision in April 2024.
The finalized guidance, titled "Promotional Labeling and Advertising Considerations for Prescription Biological Reference Products, Biosimilar Products, and Interchangeable Biosimilar Products: Questions and Answers," arrives at a pivotal moment for the biologics sector. As of the end of Q4 2024, the FDA had approved 64 biosimilars across 17 unique biological molecules, with 41 biosimilars actively marketed in the United States. This surge in biosimilar development and commercialization underscores the urgent need for regulatory clarity—both to protect patients and to foster a competitive, innovative marketplace.
The Evolution of Biosimilar Promotion: Why This Guidance Matters
The biologics market has undergone a dramatic transformation over the past decade. Once dominated by a handful of reference products with lengthy exclusivity periods, the landscape now features a growing array of biosimilars offering comparable safety, efficacy, and quality at potentially lower costs. This evolution has brought enormous promise for patients, healthcare providers, and payers, but it has also introduced new complexities—particularly in product promotion.
Historically, the absence of clear, product-specific promotional guidelines for biosimilars and their reference products created a gray area. Manufacturers, regulatory professionals, and healthcare communicators often grappled with questions about what constituted fair, accurate, and non-misleading promotional practices. The risk of inadvertently crossing regulatory lines was ever-present, with significant consequences for non-compliance.
The FDA’s finalized guidance decisively addresses these challenges. By articulating specific expectations and providing illustrative examples, the agency empowers manufacturers to confidently communicate the value of their products—while safeguarding public health and ensuring that patients and providers receive truthful, balanced information.
What the Guidance Addresses: A Framework for Clarity
At its core, the FDA’s guidance adopts a question-and-answer format, addressing nine key areas that manufacturers, packers, distributors, and their representatives routinely encounter when developing promotional communications. The central principle is unwavering: all promotional materials must be “accurate, truthful, and non-misleading” regarding a drug’s safety and effectiveness.
Product Identification Requirements
One of the most critical and often overlooked aspects of promotional compliance is precise product identification. The guidance requires firms to evaluate the context and content of each promotional communication to ensure it accurately identifies whether the message pertains to a reference product, a biosimilar, or both. This level of specificity is essential to prevent the misattribution of data or claims, which could inadvertently mislead healthcare professionals or patients.
For example, if a promotional piece references clinical data, it must be clear whether the data pertains to the reference product, the biosimilar, or both. Ambiguity in this area can have far-reaching implications, potentially undermining trust in biosimilars and impeding their adoption.
Use of Reference Product Data
The guidance also addresses the nuanced issue of referencing data from studies conducted to support the licensure of the reference product. When such data appears in both the biosimilar’s and the reference product’s FDA-approved labeling, biosimilar manufacturers are directed to cite their own product’s labeling rather than that of the reference product. This approach reinforces the principle that biosimilars are independently evaluated and approved on the basis of robust scientific evidence.
This distinction is more than a technicality—it reflects the FDA’s commitment to ensuring that promotional materials accurately represent the regulatory status and scientific foundation of each product. By requiring biosimilar manufacturers to reference their own labeling, the guidance helps prevent confusion and supports informed decision-making by healthcare providers.
Comparative Claims Between Products
Perhaps the most consequential aspect of the guidance concerns comparative advertising. The FDA takes a firm stance: once a biosimilar is licensed, the agency has determined that there are no clinically meaningful differences in safety, purity, and potency between the biosimilar and its reference product. Promotional communications that suggest otherwise—whether explicitly or implicitly—are likely to be deemed false or misleading.
This position has profound implications for marketing strategy. Manufacturers must exercise caution when crafting comparative claims, ensuring that any statements about differences between products are grounded in robust, statistically significant evidence and do not create unwarranted impressions of superiority or inferiority.
Prohibited Promotional Practices: Drawing Clear Lines
To further clarify expectations, the FDA explicitly identifies several promotional approaches that are considered misleading and therefore prohibited.
Prohibited Claims of Superiority or Inferiority
Manufacturers are barred from suggesting that a reference product is safer or more effective than an approved biosimilar. Similarly, biosimilar makers cannot claim superiority over their reference products based on data from biosimilarity studies. The guidance also prohibits positioning one approved biosimilar as better than another for the same reference product, and it forbids implying that a biosimilar is less safe or effective simply because it lacks an interchangeability designation.
These prohibitions are grounded in the FDA’s rigorous scientific review process. Both biosimilars and interchangeable biosimilars must meet the same high standard of biosimilarity for approval. The presence or absence of an interchangeability designation does not, in itself, imply differences in safety or effectiveness.
Indications and Comparative Claims
The guidance warns against using the number of licensed indications as a basis for comparative claims. A biosimilar licensed for fewer indications than its reference product should not be portrayed as less safe or effective solely because of this difference. Such claims risk misleading healthcare providers and patients, potentially discouraging the use of safe and effective biosimilars.
Interchangeability Designation
Manufacturers cannot claim that a biosimilar lacking an interchangeability designation is inferior to one that has achieved it. The interchangeability designation is a regulatory distinction that facilitates pharmacy-level substitution in certain circumstances, but it does not confer inherent superiority in terms of safety or efficacy.
Illustrative Examples: Bringing the Guidance to Life
To help manufacturers navigate these complex requirements, the FDA provides four detailed examples using fictional products. These scenarios illustrate both compliant and non-compliant promotional practices, offering valuable insights for regulatory and marketing teams.
Acceptable Practice: Highlighting Similarities
In one example, a biosimilar manufacturer includes claims that its product has the same route of administration, dosage form, and strength as the reference product for shared licensed indications. The manufacturer also states that healthcare providers can consider prescribing the biosimilar to both treatment-naïve patients and those currently receiving the reference product. These claims are supported by licensure data and are presented in a balanced, non-misleading manner.
Acceptable Practice: Presenting Comparative Study Data
Another example involves a firm presenting data from a comparative clinical study that supported the demonstration of biosimilarity, even though the data is not included in the FDA-approved labeling. The presentation is deemed acceptable because it is consistent with approved labeling, includes appropriate context about study design and methodology, describes the study’s role in biosimilarity evaluation, and notes any material limitations.
Misleading Practice: Overstating Differences
In a cautionary example, promotional communications for a reference product state that patients experienced numerically higher response rates than those on a biosimilar, based on a biosimilarity study. This is misleading because the difference was not statistically significant, and the presentation implies clinically meaningful differences where none exist.
Misleading Practice: Misrepresenting Interchangeability
A final example highlights the risks of misrepresenting the significance of interchangeability. An interchangeable biosimilar’s promotional materials claim that patients can be “assured” of its safety and effectiveness because it is licensed as interchangeable, while a competing biosimilar is not. This misleadingly suggests that the interchangeable product is superior in safety and efficacy, contrary to the FDA’s scientific determinations.
Key Changes from the 2024 Draft: Embracing the Digital Age
The finalized guidance incorporates several important changes from the 2024 draft, reflecting both stakeholder feedback and the evolving realities of pharmaceutical marketing.
Medium-Agnostic Recommendations
One of the most significant clarifications is that the guidance applies regardless of the communication medium. Whether promotional materials are disseminated via print, digital channels, social media, or interactive platforms, the same standards of accuracy, truthfulness, and non-misleading content apply. This medium-agnostic approach is especially relevant as pharmaceutical marketing increasingly leverages digital and social platforms to reach diverse audiences.
Enhanced Clarity and Consistency
The final guidance also includes expanded discussion of the considerations firms should take into account when comparing biosimilar products and reference products. Editorial changes throughout the document enhance consistency, readability, and clarity, making it easier for manufacturers and regulatory professionals to interpret and apply the guidance in real-world scenarios.
Postmarketing Reporting Requirements: Ensuring Ongoing Oversight
The guidance reaffirms that postmarketing reporting requirements apply to promotional communications for both reference products and biosimilars. Manufacturers must submit specimens of mailing pieces and other promotional labeling or advertising to the FDA at the time of initial dissemination or publication, accompanied by Form FDA 2253. This requirement ensures ongoing regulatory oversight and supports the FDA’s ability to monitor promotional practices in real time.
Market Context and Industry Implications: A New Era of Competition
The FDA’s regulatory clarity arrives at a time of unprecedented growth and transformation in the biosimilar market. In 2025, the global biosimilar market is valued at USD 41.97 billion and is projected to reach USD 97.32 billion by 203, expanding at a robust compound annual growth rate (CAGR) of 18.32%. This explosive growth is driven by a confluence of factors, including the expiration of exclusivity for major biologics, advances in manufacturing technology, and increasing acceptance of biosimilars among healthcare providers and patients.
The Stakes for Promotional Compliance
The stakes for promotional compliance are higher than ever. More than USD 170 billion in biologic revenues are expected to lose exclusivity by 203, creating intense competitive pressure between reference product manufacturers seeking to retain market share and biosimilar developers seeking to capture it. In this environment, effective—and compliant—promotional strategies are essential for success.
Market Penetration: Lessons from Oncology and Beyond
The experience of biosimilars in oncology, ophthalmology, and pegfilgrastim provides a compelling case study. These products achieved an average biosimilar market share of 81% within five years of launch, demonstrating that biosimilars can achieve substantial market penetration when effectively commercialized. In contrast, biosimilars in immunology, filgrastim, epoetin alfa, and insulin glargine achieved only a 26% average market share after five years, highlighting the critical role of promotional strategies and market dynamics in driving adoption.
The FDA’s Commitment to Truthful Promotion
The FDA has consistently signaled its commitment to addressing misleading biosimilar promotion. The agency has taken action against false and misleading communications, including inappropriate comparisons between biosimilars and their reference products. The finalized guidance builds on this foundation, providing manufacturers with the tools they need to navigate the complex regulatory landscape with confidence.
Compliance Review in the Digital Age: Harnessing AI for Regulatory Excellence
For compliance professionals, the FDA’s detailed guidance presents both an opportunity and a challenge. On the one hand, the specificity of the requirements enables a more objective compliance assessment. On the other hand, the sheer volume of promotional content—spanning multiple channels and formats—demands efficient, scalable review processes.
AI-Powered Compliance Review: A Transformative Solution
Enter AI-powered compliance review systems, such as Signify, which are revolutionizing how regulatory teams review promotional materials. These systems leverage advanced natural language processing, machine learning, and data analytics to streamline the compliance review process, ensuring that promotional materials align with FDA guidance while freeing up human reviewers to focus on higher-level analysis and judgment.
Step 1: Document Ingestion and Requirement Extraction
The AI system begins by parsing the FDA guidance to identify discrete compliance requirements, including the nine Q&A areas, specific prohibited claims, and illustrative examples. This process creates a structured checklist against which promotional materials can be evaluated, ensuring comprehensive coverage of all relevant regulatory provisions.

Step 2: Promotional Material Analysis
When a draft promotional piece is submitted for review, the system analyzes the content to identify all product claims, comparative statements, data citations, and implied messages. Natural language processing algorithms flag statements that could suggest superiority, inferiority, or clinically meaningful differences, enabling early detection of potential compliance issues.
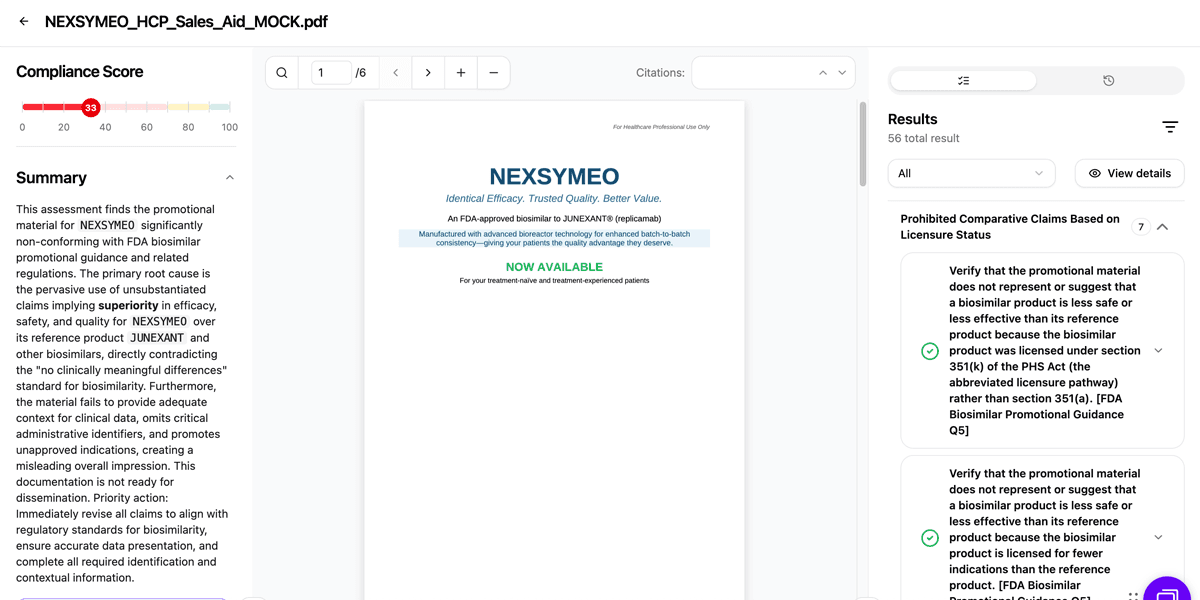
Step 3: Cross-Reference Validation
The system cross-references data cited in promotional materials with the product’s FDA-approved labeling, verifying that reference product data is properly attributed and that product identification (proprietary name, proper name, core name) meets the guidance’s specificity requirements. This automated validation reduces the risk of inadvertent misattribution or ambiguity.
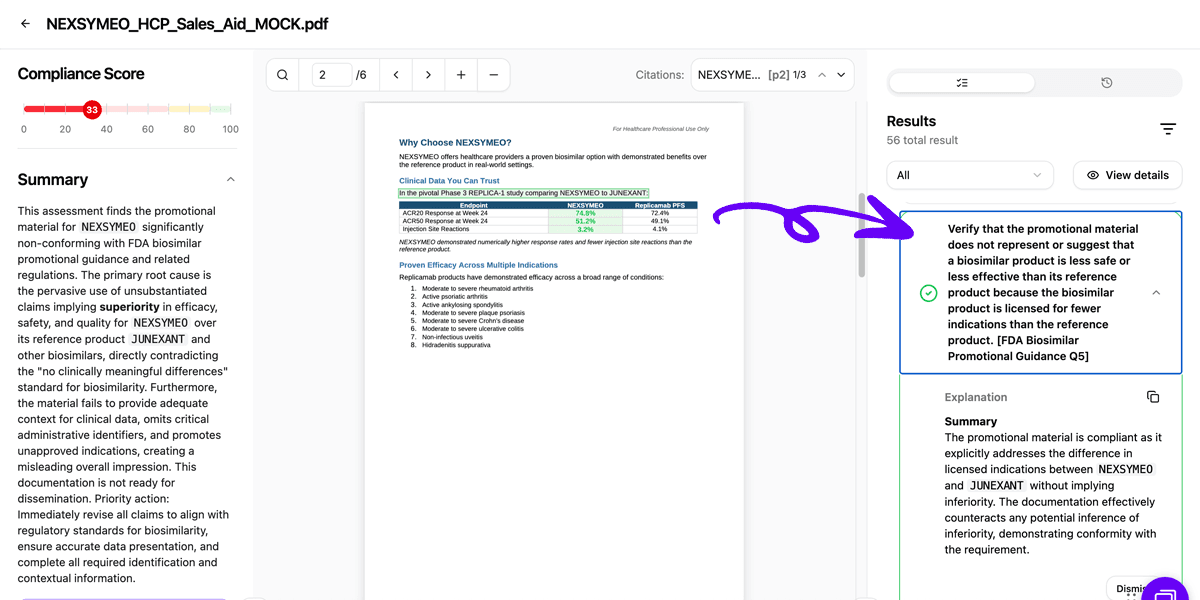
Step 4: Risk Flagging and Categorization
Potential compliance issues are categorized by severity. High-risk flags include explicit superiority claims between biosimilars, suggestions that non-interchangeable biosimilars are less safe or effective, or comparative efficacy claims based on non-statistically significant differences. Medium-risk flags might include ambiguous comparative language or incomplete study context, while low-risk flags address formatting issues or minor labeling inconsistencies.
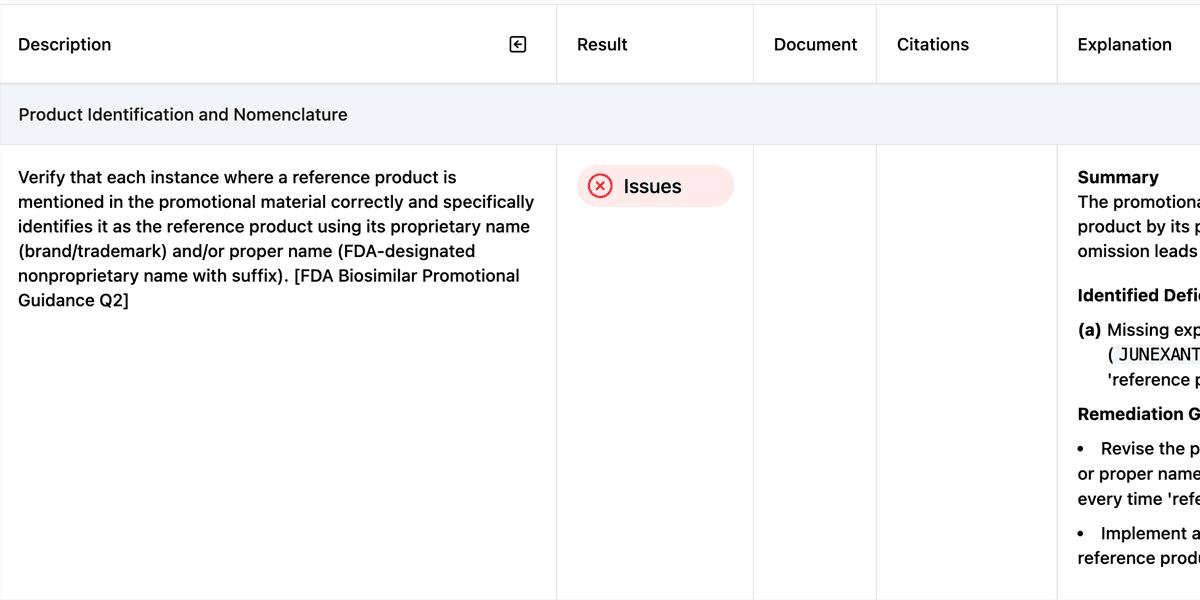
Step 5: Remediation Recommendations
For each flagged issue, the system provides specific guidance, language citations, and suggests compliant alternative phrasing. For example, if promotional copy states that a reference product has “proven efficacy over 15 years of clinical use” in a comparative context with a newer biosimilar, the system would flag this as potentially implying the biosimilar lacks equivalent efficacy and recommend neutral language that avoids misleading impressions.

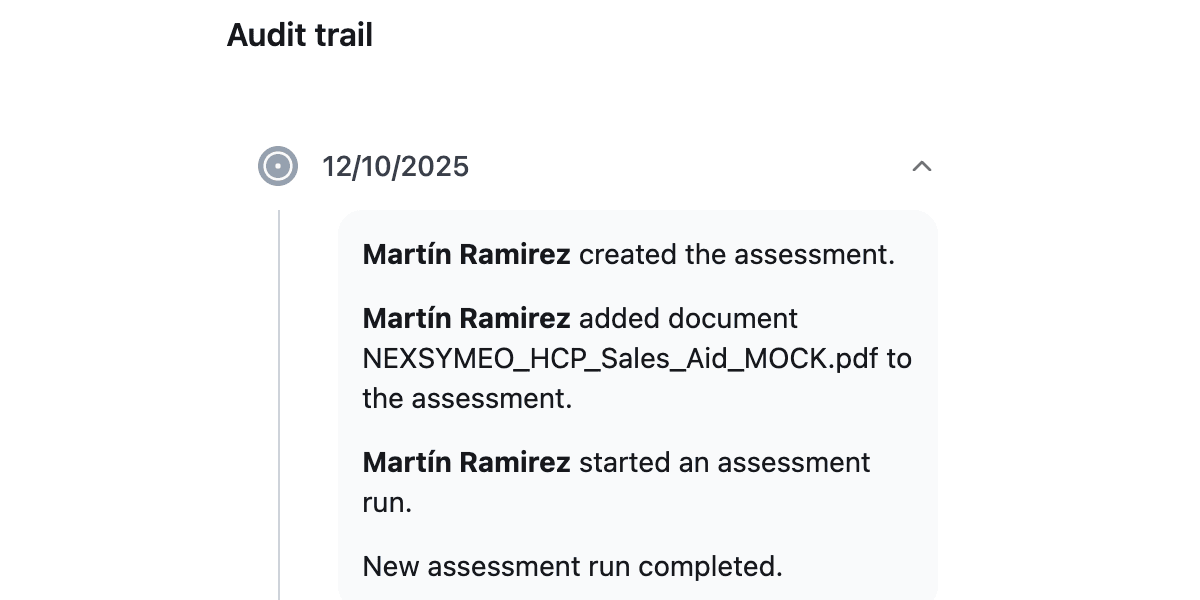
Step 6: Audit Trail Documentation
The review process generates a comprehensive compliance report documenting each identified issue, the specific guidance provision implicated, and the resolution status. This documentation supports both internal quality processes and FDA inspection readiness, providing a transparent audit trail of compliance efforts.
The Human Element: Augmenting, Not Replacing, Expertise
While AI-powered systems offer significant efficiency gains, they are designed to augment—not replace—the expertise of regulatory professionals. Human judgment remains essential for context-dependent determinations, nuanced interpretation of guidance, and the development of creative, compliant promotional strategies. By automating routine tasks and flagging potential issues, AI empowers compliance teams to focus on higher-value activities that drive business success and protect public health.
Looking Ahead: Building a Foundation for the Future
The FDA’s finalized guidance marks the culmination of the agency’s commitment, under the Biosimilar User Fee Amendments of 2022 (BsUFA III), to provide clarity on promotional communications for interchangeable biosimilar products. With this regulatory framework now firmly in place, manufacturers have more precise parameters for competitive marketing that support both business objectives and the FDA’s public health mission.
The Road Ahead for Manufacturers
As the biologics market continues to evolve, with new biosimilar launches for high-value molecules such as ustekinumab, aflibercept, and denosumab on the horizon, this guidance will serve as the foundational reference for promotional compliance across the industry. Manufacturers that embrace the guidance’s principles—accuracy, truthfulness, and non-misleading communication—will be well-positioned to build trust with healthcare providers, patients, and payers.
Empowering Healthcare Providers and Patients
Ultimately, the FDA’s guidance is about more than regulatory compliance—it is about empowering healthcare providers and patients to make informed decisions. By ensuring that promotional communications are grounded in science and free from misleading claims, the guidance supports the broader goal of expanding access to safe, effective, and affordable biologic therapies.
A Call to Action: Embracing Compliance as a Competitive Advantage
In today’s dynamic and competitive biologics market, compliance is not a burden—it is a strategic advantage. Manufacturers that invest in robust compliance processes, leverage advanced technologies, and foster a culture of integrity will not only avoid regulatory pitfalls but also differentiate themselves as trusted partners in the healthcare ecosystem.
The path forward is clear. By aligning promotional practices with the FDA’s guidance, manufacturers can confidently navigate the complexities of the biosimilar market, drive sustainable growth, and contribute to a healthier, more equitable future for all.
The complete FDA guidance document is available through the FDA’s guidance database and the Federal Register. Firms seeking FDA feedback on draft promotional communications before dissemination should follow the established process outlined in 21 CFR 202.1(j)(4).
References
U.S. Food and Drug Administration. “Promotional Labeling and Advertising Considerations for Prescription Biological Reference Products, Biosimilar Products, and Interchangeable Biosimilar Products: Questions and Answers.” Federal Register, December 10, 2025.
FDA Guidance Database. [Link]
Market research reports on biosimilar market size and forecasts.
Industry analyses on biosimilar adoption and market penetration.
BsUFA III legislative documents.
Regulatory Standards in Biosimilar Promotional Labeling: What You Need to Know
In a landmark move for the biologics industry, the U.S. Food and Drug Administration (FDA) has finalized its long-awaited guidance on promotional labeling and advertising for prescription biological products. This comprehensive document establishes clear, enforceable boundaries for how manufacturers can market biosimilars, interchangeable biosimilars, and their reference products. Published in the Federal Register on December 10, 2025, the guidance marks the culmination of a regulatory journey that began with draft guidance in 202 and underwent significant revision in April 2024.
The finalized guidance, titled "Promotional Labeling and Advertising Considerations for Prescription Biological Reference Products, Biosimilar Products, and Interchangeable Biosimilar Products: Questions and Answers," arrives at a pivotal moment for the biologics sector. As of the end of Q4 2024, the FDA had approved 64 biosimilars across 17 unique biological molecules, with 41 biosimilars actively marketed in the United States. This surge in biosimilar development and commercialization underscores the urgent need for regulatory clarity—both to protect patients and to foster a competitive, innovative marketplace.
The Evolution of Biosimilar Promotion: Why This Guidance Matters
The biologics market has undergone a dramatic transformation over the past decade. Once dominated by a handful of reference products with lengthy exclusivity periods, the landscape now features a growing array of biosimilars offering comparable safety, efficacy, and quality at potentially lower costs. This evolution has brought enormous promise for patients, healthcare providers, and payers, but it has also introduced new complexities—particularly in product promotion.
Historically, the absence of clear, product-specific promotional guidelines for biosimilars and their reference products created a gray area. Manufacturers, regulatory professionals, and healthcare communicators often grappled with questions about what constituted fair, accurate, and non-misleading promotional practices. The risk of inadvertently crossing regulatory lines was ever-present, with significant consequences for non-compliance.
The FDA’s finalized guidance decisively addresses these challenges. By articulating specific expectations and providing illustrative examples, the agency empowers manufacturers to confidently communicate the value of their products—while safeguarding public health and ensuring that patients and providers receive truthful, balanced information.
What the Guidance Addresses: A Framework for Clarity
At its core, the FDA’s guidance adopts a question-and-answer format, addressing nine key areas that manufacturers, packers, distributors, and their representatives routinely encounter when developing promotional communications. The central principle is unwavering: all promotional materials must be “accurate, truthful, and non-misleading” regarding a drug’s safety and effectiveness.
Product Identification Requirements
One of the most critical and often overlooked aspects of promotional compliance is precise product identification. The guidance requires firms to evaluate the context and content of each promotional communication to ensure it accurately identifies whether the message pertains to a reference product, a biosimilar, or both. This level of specificity is essential to prevent the misattribution of data or claims, which could inadvertently mislead healthcare professionals or patients.
For example, if a promotional piece references clinical data, it must be clear whether the data pertains to the reference product, the biosimilar, or both. Ambiguity in this area can have far-reaching implications, potentially undermining trust in biosimilars and impeding their adoption.
Use of Reference Product Data
The guidance also addresses the nuanced issue of referencing data from studies conducted to support the licensure of the reference product. When such data appears in both the biosimilar’s and the reference product’s FDA-approved labeling, biosimilar manufacturers are directed to cite their own product’s labeling rather than that of the reference product. This approach reinforces the principle that biosimilars are independently evaluated and approved on the basis of robust scientific evidence.
This distinction is more than a technicality—it reflects the FDA’s commitment to ensuring that promotional materials accurately represent the regulatory status and scientific foundation of each product. By requiring biosimilar manufacturers to reference their own labeling, the guidance helps prevent confusion and supports informed decision-making by healthcare providers.
Comparative Claims Between Products
Perhaps the most consequential aspect of the guidance concerns comparative advertising. The FDA takes a firm stance: once a biosimilar is licensed, the agency has determined that there are no clinically meaningful differences in safety, purity, and potency between the biosimilar and its reference product. Promotional communications that suggest otherwise—whether explicitly or implicitly—are likely to be deemed false or misleading.
This position has profound implications for marketing strategy. Manufacturers must exercise caution when crafting comparative claims, ensuring that any statements about differences between products are grounded in robust, statistically significant evidence and do not create unwarranted impressions of superiority or inferiority.
Prohibited Promotional Practices: Drawing Clear Lines
To further clarify expectations, the FDA explicitly identifies several promotional approaches that are considered misleading and therefore prohibited.
Prohibited Claims of Superiority or Inferiority
Manufacturers are barred from suggesting that a reference product is safer or more effective than an approved biosimilar. Similarly, biosimilar makers cannot claim superiority over their reference products based on data from biosimilarity studies. The guidance also prohibits positioning one approved biosimilar as better than another for the same reference product, and it forbids implying that a biosimilar is less safe or effective simply because it lacks an interchangeability designation.
These prohibitions are grounded in the FDA’s rigorous scientific review process. Both biosimilars and interchangeable biosimilars must meet the same high standard of biosimilarity for approval. The presence or absence of an interchangeability designation does not, in itself, imply differences in safety or effectiveness.
Indications and Comparative Claims
The guidance warns against using the number of licensed indications as a basis for comparative claims. A biosimilar licensed for fewer indications than its reference product should not be portrayed as less safe or effective solely because of this difference. Such claims risk misleading healthcare providers and patients, potentially discouraging the use of safe and effective biosimilars.
Interchangeability Designation
Manufacturers cannot claim that a biosimilar lacking an interchangeability designation is inferior to one that has achieved it. The interchangeability designation is a regulatory distinction that facilitates pharmacy-level substitution in certain circumstances, but it does not confer inherent superiority in terms of safety or efficacy.
Illustrative Examples: Bringing the Guidance to Life
To help manufacturers navigate these complex requirements, the FDA provides four detailed examples using fictional products. These scenarios illustrate both compliant and non-compliant promotional practices, offering valuable insights for regulatory and marketing teams.
Acceptable Practice: Highlighting Similarities
In one example, a biosimilar manufacturer includes claims that its product has the same route of administration, dosage form, and strength as the reference product for shared licensed indications. The manufacturer also states that healthcare providers can consider prescribing the biosimilar to both treatment-naïve patients and those currently receiving the reference product. These claims are supported by licensure data and are presented in a balanced, non-misleading manner.
Acceptable Practice: Presenting Comparative Study Data
Another example involves a firm presenting data from a comparative clinical study that supported the demonstration of biosimilarity, even though the data is not included in the FDA-approved labeling. The presentation is deemed acceptable because it is consistent with approved labeling, includes appropriate context about study design and methodology, describes the study’s role in biosimilarity evaluation, and notes any material limitations.
Misleading Practice: Overstating Differences
In a cautionary example, promotional communications for a reference product state that patients experienced numerically higher response rates than those on a biosimilar, based on a biosimilarity study. This is misleading because the difference was not statistically significant, and the presentation implies clinically meaningful differences where none exist.
Misleading Practice: Misrepresenting Interchangeability
A final example highlights the risks of misrepresenting the significance of interchangeability. An interchangeable biosimilar’s promotional materials claim that patients can be “assured” of its safety and effectiveness because it is licensed as interchangeable, while a competing biosimilar is not. This misleadingly suggests that the interchangeable product is superior in safety and efficacy, contrary to the FDA’s scientific determinations.
Key Changes from the 2024 Draft: Embracing the Digital Age
The finalized guidance incorporates several important changes from the 2024 draft, reflecting both stakeholder feedback and the evolving realities of pharmaceutical marketing.
Medium-Agnostic Recommendations
One of the most significant clarifications is that the guidance applies regardless of the communication medium. Whether promotional materials are disseminated via print, digital channels, social media, or interactive platforms, the same standards of accuracy, truthfulness, and non-misleading content apply. This medium-agnostic approach is especially relevant as pharmaceutical marketing increasingly leverages digital and social platforms to reach diverse audiences.
Enhanced Clarity and Consistency
The final guidance also includes expanded discussion of the considerations firms should take into account when comparing biosimilar products and reference products. Editorial changes throughout the document enhance consistency, readability, and clarity, making it easier for manufacturers and regulatory professionals to interpret and apply the guidance in real-world scenarios.
Postmarketing Reporting Requirements: Ensuring Ongoing Oversight
The guidance reaffirms that postmarketing reporting requirements apply to promotional communications for both reference products and biosimilars. Manufacturers must submit specimens of mailing pieces and other promotional labeling or advertising to the FDA at the time of initial dissemination or publication, accompanied by Form FDA 2253. This requirement ensures ongoing regulatory oversight and supports the FDA’s ability to monitor promotional practices in real time.
Market Context and Industry Implications: A New Era of Competition
The FDA’s regulatory clarity arrives at a time of unprecedented growth and transformation in the biosimilar market. In 2025, the global biosimilar market is valued at USD 41.97 billion and is projected to reach USD 97.32 billion by 203, expanding at a robust compound annual growth rate (CAGR) of 18.32%. This explosive growth is driven by a confluence of factors, including the expiration of exclusivity for major biologics, advances in manufacturing technology, and increasing acceptance of biosimilars among healthcare providers and patients.
The Stakes for Promotional Compliance
The stakes for promotional compliance are higher than ever. More than USD 170 billion in biologic revenues are expected to lose exclusivity by 203, creating intense competitive pressure between reference product manufacturers seeking to retain market share and biosimilar developers seeking to capture it. In this environment, effective—and compliant—promotional strategies are essential for success.
Market Penetration: Lessons from Oncology and Beyond
The experience of biosimilars in oncology, ophthalmology, and pegfilgrastim provides a compelling case study. These products achieved an average biosimilar market share of 81% within five years of launch, demonstrating that biosimilars can achieve substantial market penetration when effectively commercialized. In contrast, biosimilars in immunology, filgrastim, epoetin alfa, and insulin glargine achieved only a 26% average market share after five years, highlighting the critical role of promotional strategies and market dynamics in driving adoption.
The FDA’s Commitment to Truthful Promotion
The FDA has consistently signaled its commitment to addressing misleading biosimilar promotion. The agency has taken action against false and misleading communications, including inappropriate comparisons between biosimilars and their reference products. The finalized guidance builds on this foundation, providing manufacturers with the tools they need to navigate the complex regulatory landscape with confidence.
Compliance Review in the Digital Age: Harnessing AI for Regulatory Excellence
For compliance professionals, the FDA’s detailed guidance presents both an opportunity and a challenge. On the one hand, the specificity of the requirements enables a more objective compliance assessment. On the other hand, the sheer volume of promotional content—spanning multiple channels and formats—demands efficient, scalable review processes.
AI-Powered Compliance Review: A Transformative Solution
Enter AI-powered compliance review systems, such as Signify, which are revolutionizing how regulatory teams review promotional materials. These systems leverage advanced natural language processing, machine learning, and data analytics to streamline the compliance review process, ensuring that promotional materials align with FDA guidance while freeing up human reviewers to focus on higher-level analysis and judgment.
Step 1: Document Ingestion and Requirement Extraction
The AI system begins by parsing the FDA guidance to identify discrete compliance requirements, including the nine Q&A areas, specific prohibited claims, and illustrative examples. This process creates a structured checklist against which promotional materials can be evaluated, ensuring comprehensive coverage of all relevant regulatory provisions.
Step 2: Promotional Material Analysis
When a draft promotional piece is submitted for review, the system analyzes the content to identify all product claims, comparative statements, data citations, and implied messages. Natural language processing algorithms flag statements that could suggest superiority, inferiority, or clinically meaningful differences, enabling early detection of potential compliance issues.
Step 3: Cross-Reference Validation
The system cross-references data cited in promotional materials with the product’s FDA-approved labeling, verifying that reference product data is properly attributed and that product identification (proprietary name, proper name, core name) meets the guidance’s specificity requirements. This automated validation reduces the risk of inadvertent misattribution or ambiguity.
Step 4: Risk Flagging and Categorization
Potential compliance issues are categorized by severity. High-risk flags include explicit superiority claims between biosimilars, suggestions that non-interchangeable biosimilars are less safe or effective, or comparative efficacy claims based on non-statistically significant differences. Medium-risk flags might include ambiguous comparative language or incomplete study context, while low-risk flags address formatting issues or minor labeling inconsistencies.
Step 5: Remediation Recommendations
For each flagged issue, the system provides specific guidance, language citations, and suggests compliant alternative phrasing. For example, if promotional copy states that a reference product has “proven efficacy over 15 years of clinical use” in a comparative context with a newer biosimilar, the system would flag this as potentially implying the biosimilar lacks equivalent efficacy and recommend neutral language that avoids misleading impressions.
Step 6: Audit Trail Documentation
The review process generates a comprehensive compliance report documenting each identified issue, the specific guidance provision implicated, and the resolution status. This documentation supports both internal quality processes and FDA inspection readiness, providing a transparent audit trail of compliance efforts.
The Human Element: Augmenting, Not Replacing, Expertise
While AI-powered systems offer significant efficiency gains, they are designed to augment—not replace—the expertise of regulatory professionals. Human judgment remains essential for context-dependent determinations, nuanced interpretation of guidance, and the development of creative, compliant promotional strategies. By automating routine tasks and flagging potential issues, AI empowers compliance teams to focus on higher-value activities that drive business success and protect public health.
Looking Ahead: Building a Foundation for the Future
The FDA’s finalized guidance marks the culmination of the agency’s commitment, under the Biosimilar User Fee Amendments of 2022 (BsUFA III), to provide clarity on promotional communications for interchangeable biosimilar products. With this regulatory framework now firmly in place, manufacturers have more precise parameters for competitive marketing that support both business objectives and the FDA’s public health mission.
The Road Ahead for Manufacturers
As the biologics market continues to evolve, with new biosimilar launches for high-value molecules such as ustekinumab, aflibercept, and denosumab on the horizon, this guidance will serve as the foundational reference for promotional compliance across the industry. Manufacturers that embrace the guidance’s principles—accuracy, truthfulness, and non-misleading communication—will be well-positioned to build trust with healthcare providers, patients, and payers.
Empowering Healthcare Providers and Patients
Ultimately, the FDA’s guidance is about more than regulatory compliance—it is about empowering healthcare providers and patients to make informed decisions. By ensuring that promotional communications are grounded in science and free from misleading claims, the guidance supports the broader goal of expanding access to safe, effective, and affordable biologic therapies.
A Call to Action: Embracing Compliance as a Competitive Advantage
In today’s dynamic and competitive biologics market, compliance is not a burden—it is a strategic advantage. Manufacturers that invest in robust compliance processes, leverage advanced technologies, and foster a culture of integrity will not only avoid regulatory pitfalls but also differentiate themselves as trusted partners in the healthcare ecosystem.
The path forward is clear. By aligning promotional practices with the FDA’s guidance, manufacturers can confidently navigate the complexities of the biosimilar market, drive sustainable growth, and contribute to a healthier, more equitable future for all.
The complete FDA guidance document is available through the FDA’s guidance database and the Federal Register. Firms seeking FDA feedback on draft promotional communications before dissemination should follow the established process outlined in 21 CFR 202.1(j)(4).
References
U.S. Food and Drug Administration. “Promotional Labeling and Advertising Considerations for Prescription Biological Reference Products, Biosimilar Products, and Interchangeable Biosimilar Products: Questions and Answers.” Federal Register, December 10, 2025.
FDA Guidance Database. [Link]
Market research reports on biosimilar market size and forecasts.
Industry analyses on biosimilar adoption and market penetration.
BsUFA III legislative documents.
The information presented is for educational and informational purposes only and should not be construed as legal, regulatory, or professional advice. Organizations should consult with qualified legal and compliance professionals for guidance specific to their circumstances.
Biosimilar Promotional Labeling Guidance Is Final
Dec 10, 2025